






Síndrome de Kallmann

Content
sinais e sintomas

Normalmente, é difícil distinguir um caso de síndrome de Kallmann (KS)/hipogonadismo hipogotrópico (HH) a partir de um atraso constitucional direto da puberdade. No entanto, se a puberdade não tiver começado aos 14 anos (meninas) ou 15 (meninos) anos e um ou mais dos recursos não reprodutivos mencionados abaixo estão presentes, um encaminhamento para o endocrinologista reprodutivo pode ser aconselhável.
As características do KS e outras formas de HH podem ser divididas em duas categorias diferentes; "reprodutivo" e "não reprodutivo".
Características reprodutivas
Failure to start or fully complete puberty.Lack of testicle development in men (size < 4 ml, whereas the normal range is between 12 and 25 ml).Primary amenorrhoea (failure to start menstruation).Poorly defined secondary sexual characteristics.Micropenis in 5-10% of male cases.Cryptorchidism (undescended testicles) at birth.Low levels of the gonadotropins LH and FSH.Hypogonadism due to low levels of testosterone in men or oestrogen/progesterone in women.Infertility.Recursos não reprodutivos
Total lack of sense of smell (anosmia) or markedly reduced sense of smell (hyposmia). This is the defining feature of Kallmann syndrome; it is not seen in other cases of HH. Approximately 50% of HH cases occur with anosmia and can be termed as Kallmann syndrome.Cleft palate, cleft lip or other midline cranio-facial defects.Neural hearing impairmentAbsence of one of the kidneys (unilateral renal agenesis)Skeletal defects including split hand/foot (ectrodactyly), shortened middle finger (metacarpal) or scoliosisManual synkinesis (mirror movements of hands)Missing teeth (hypodontia)Poor balance or coordination due to cerebral ataxia.Eye defects such as coloboma or ptosis.Increased incidence of color-blindnessA natureza genética exata de cada caso específico de KS/HH determinará quais, se houver, das características não reprodutivas ocorrerão. A gravidade dos sintomas também variará de caso para caso. Até os membros da família não mostram o mesmo alcance ou gravidade dos sintomas.
O KS/HH está frequentemente presente desde o nascimento, mas as versões de início de adultos são encontradas em homens e mulheres. O eixo hipotalâmico-hipófise-gonadal (eixo HPG) funciona normalmente no nascimento e bem na vida adulta, dando puberdade normal e função reprodutiva normal. O eixo HPG falha totalmente ou é reduzido a um nível muito baixo de liberação de GnRH na vida adulta sem causa óbvia (por exemplo, um tumor hipofisário). Isso levará a uma queda nos níveis de testosterona ou estrogênio e infertilidade.
A amenorréia hipotalâmica funcional é vista nas fêmeas onde o eixo HPG é suprimido em resposta ao estresse físico ou psicológico ou desnutrição, mas é reversível com a remoção do estressor.
Alguns casos de Ks/Hh parecem reverter durante a vida adulta, onde o eixo HPG retoma sua função normal e os níveis de GnRH, LH e FSH retornam aos níveis normais. Isso ocorre em cerca de 10 a 22% das pessoas, principalmente casos de ChH normosmicos, em vez de casos de KS e apenas encontrados em pessoas submetidas a alguma forma de terapia de reposição de testosterona. É normalmente descoberto quando o volume testicular aumenta enquanto no tratamento com testosterona sozinho e os níveis de testosterona retornam ao normal quando o tratamento é interrompido. Esse tipo de ks/hh raramente ocorre nos casos em que os homens tiveram um histórico de testículos não descendentes.
Indivíduos afetados com KS e outras formas de HH são quase invariavelmente nascidos com diferenciação sexual normal; isto é, eles são fisicamente masculinos ou femininos. Isso ocorre devido à gonadotrofina coriônica humana (HCG) produzida pela placenta em aproximadamente 12 a 20 semanas de gestação (gravidez), que normalmente não é afetada por KS ou CHH.
Pessoas com KS/HH carecem da onda de GnRH, LH e FSH que normalmente ocorre entre o nascimento e os seis meses de idade. Essa onda é particularmente importante em meninos, pois ajuda com a descida testicular no escroto. A onda de GnRH/LH/FSH em crianças não KS/HH fornece níveis detectáveis de testosterona em meninos e estrogênio e progesterona em meninas. Às vezes, a falta desse aumento pode ser usada como uma ferramenta de diagnóstico se KS/HH for suspeita de um garoto recém -nascido, mas normalmente não for distinto o suficiente para o diagnóstico em meninas.
Osteoporose
Um possível efeito colateral de ter KS/CHH é o aumento do risco de desenvolver osteoporose secundária ou osteopenia. O estrogênio (fêmeas) ou testosterona (homens) é essencial para manter a densidade óssea. A deficiência de testosterona ou estrogênio pode aumentar a taxa de reabsorção óssea e, ao mesmo tempo, diminuir a taxa de formação óssea. No geral, isso pode levar a ossos fracos e frágeis que têm uma tendência maior de fraturar. [Citação necessária]
Mesmo pouco tempo com baixo estrogênio ou testosterona, como nos casos de diagnóstico tardio de KS/CHH pode levar a um risco aumentado de desenvolver osteoporose, mas outros fatores de risco, como o tabagismo estão envolvidos, de modo que o risco de desenvolver isso variará de pessoa para pessoa. Recomenda -se as varreduras de densidade óssea para monitorar a densidade mineral óssea.
A varredura de densidade óssea é conhecida como varredura de absorciometria de raios X dupla de energia (DEXA ou DXA). É um teste simples, levando menos de 15 minutos para serem executados. Envolve tirar uma imagem especializada de raios-X da coluna e dos quadris e medir a densidade mineral óssea e comparar o resultado com o valor médio de um adulto jovem e saudável na população em geral.
Níveis adequados de cálcio e, provavelmente, mais importante, os níveis de vitamina D são essenciais para a densidade óssea saudável. Algumas pessoas com KS/CHH terão seus níveis verificados e podem receber comprimidos ou injeções extras de vitamina D para tentar impedir que a condição piorasse. O papel da vitamina D para a saúde geral geral está sob estreita escrutínio no momento, com alguns pesquisadores alegando que a deficiência de vitamina D prevalece em muitas populações e pode estar ligada a outras doenças.
Algumas pessoas com osteoporose grave podem receber bisfosfonatos prescritos para preservar a massa óssea, além da terapia de reposição hormonal.
Genética
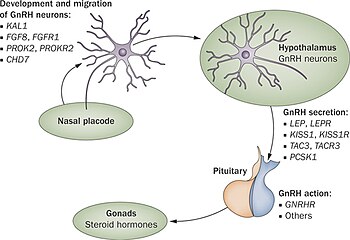
Até o momento, pelo menos 25 genes diferentes foram implicados em causar a síndrome de Kallmann ou outras formas de hipogonadismo hipogotrópico por meio de uma interrupção na produção ou atividade da GnRH (37). Esses genes envolvidos cobrem todas as formas de herança e nenhum defeito no gene demonstrou ser comum a todos os casos que dificultam o teste genético e a previsão de herança.
O número de genes conhecidos por causar casos de KS/CHH ainda está aumentando. Além disso, pensa -se que alguns casos de KS/CHH são causados por dois defeitos genéticos separados que ocorrem ao mesmo tempo.
Defeitos gênicos individuais podem estar associados a sintomas específicos que podem ajudar a identificar quais genes testarem. Entre 35 e 45% dos casos de KS/CHH têm uma causa genética desconhecida.
O defeito do gene ANOS1 (anteriormente conhecido como KAL-1) foi o primeiro descoberto e o mais comumente testado. Causa a forma ligada ao X da síndrome de Kallmann e está associada aos sintomas adicionais de anosmia, sinkinese bimanual e agenesia renal. Pensa -se que esse defeito seja responsável entre 5 e 10% de todos os casos de Síndrome de Kallmann/CHH.
Fisiopatologia
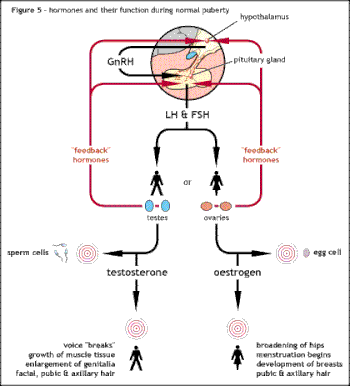

A causa subjacente da síndrome de Kallmann ou outras formas de hipogonadismo hipogonadotrópico é uma falha na ação correta do hormônio hipotalâmico GnRH. O termo deficiência isolada de GNRH (IGD) tem sido cada vez mais usada para descrever esse grupo de condições, pois destaca a principal causa dessas condições e as distingue de outras condições, como a síndrome de Klinefelter ou a síndrome de Turner, que compartilham alguns sintomas semelhantes, mas têm uma etiologia diferente . O termo hipogonadismo descreve um baixo nível de hormônios sexuais circulantes; testosterona em homens e estrogênio e progesterona em mulheres. O hipogonadismo pode ocorrer através de vários mecanismos diferentes. O uso do termo hipogonadotrópico refere -se ao fato de que o hipogonadismo encontrado no HH é causado por uma interrupção na produção dos hormônios da gonadotrofina normalmente liberados pela glândula pituitária anterior conhecida como hormônio luteinizante (LH) e follículo que estimula o hormônio (FSH). A falha na atividade da GnRH pode ser devida à ausência dos neurônios liberando GnRH dentro do hipotálamo. O HH pode ocorrer como uma condição isolada, com apenas a produção de LH e FSH sendo afetada ou pode ocorrer em condições combinadas de deficiência da hipófise. [Citação necessária]
Nas primeiras 10 semanas de desenvolvimento embrionário normal, os neurônios que liberam o GNRH migram de sua fonte original na região nasal e acabam dentro do hipotálamo. Esses neurônios se originam em uma área da cabeça em desenvolvimento, o placode olfativo, que dará origem ao epitélio olfativo; Eles então passam pela placa cribriforme, junto com as fibras dos nervos olfativos, e para o cérebro rostral. De lá, eles migram para o que se tornará o hipotálamo. Quaisquer problemas com o desenvolvimento das fibras nervosas olfativas impedirão a progressão dos neurônios liberando GnRH em direção ao cérebro.
Diagnóstico
O diagnóstico de Ks e outras formas de CHH é complicado pelas dificuldades em distinguir entre um atraso constitucional normal da puberdade ou um caso de KS/CHH. O diagnóstico é frequentemente de exclusão encontrada durante a elaboração da puberdade atrasada.
Nos homens, o uso de níveis apropriados de testosterona pode ajudar a distinguir entre um caso de KS/CHH de um caso de puberdade tardia. Se nenhuma puberdade for aparente, especialmente sem desenvolvimento testicular, uma revisão de um endocrinologista reprodutiva pode ser apropriada. Se a puberdade não é aparente aos 16 anos, a pessoa deve ser encaminhada para a revisão endocrinológica. O diagnóstico pós -natal de KS/CHH antes dos 6 meses de idade às vezes é possível, pois a onda hormonal pós -natal normal de gonadotrofinas junto com testosterona ou estrogênio está ausente em bebês com KS/CHH. Essa falta de hormônios detectáveis no sangue pode ser usada como um indicador de diagnóstico, especialmente em bebês do sexo masculino.
Nas mulheres, o diagnóstico às vezes é atrasado à medida que outras causas de amenorréia normalmente precisam ser investigadas primeiro antes que um caso de KS/CHH seja considerado.
O diagnóstico do Normal KS/CHH envolve uma série de testes clínicos, bioquímicos e radiológicos para excluir outras condições que podem causar sintomas semelhantes. [Citação necessária]
Testes clínicos
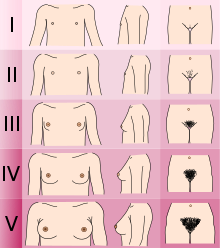
Comparing height to standard growth charts.Determining the Tanner stage of sexual development. (Males with KS/CHH are normally at stage I or II with genitalia, females at stage I with breast development and both males and females at stage III with pubic hair development).Checking for micropenis and undescended testes (cryptorchidism) in males.Measuring testicular volume.Checking for breast development and age at menarche in females.Checking sense of smell using odorant panel or University of Pennsylvania Smell Identification Test (UPSIT)Checking for hearing impairment.Checking for missing teeth or presence of cleft lip and/or cleft palate.Checking for pigmentation of skin and hair.Checking for mirror movements of the hands or signs of neurodevelopmental delay.Testes de laboratório
Early morning hormonal testing including FSH, LH, testosterone, oestrogen and prolactin.GnRH and/or hCG stimulation test to determine activity of hypothalamus and pituitary.Sperm testLiver function, renal function and inflammation marker testing.Karyotype to check for chromosomal abnormalities.Imagem médica
Performing wrist x-ray to determine bone age.Brain MRI to rule out any structural abnormalities in the hypothalamus or pituitary and to check for presence of olfactory bulbs.Ultrasound of kidneys to rule out unilateral renal agenesis.Bone density scan (DXA) to check for osteoporosis or osteopenia.Tratamento
Para homens e mulheres, o objetivo inicial do tratamento é o desenvolvimento das características sexuais secundárias normalmente observadas na puberdade. Uma vez que isso foi alcançado, a terapia contínua de reposição hormonal é necessária para machos e fêmeas manterem a função sexual, a saúde óssea, a libido e o bem -estar geral. Nos machos, a terapia de reposição de testosterona é necessária para a manutenção da massa muscular normal.
Às vezes, é necessário tratamento precoce para bebês do sexo masculino com suspeita de KS/CHH para corrigir testículos e micropénis não descendentes se apresentarem uso ou cirurgia ou gonadotrofina ou tratamento com DHT. As fêmeas com KS/CHH normalmente não requerem tratamento antes da adolescência. Atualmente, não existem tratamentos para a falta de olfato, o movimento espelhado das mãos ou a ausência de um rim.
O tratamento para homens e mulheres com KS/CHH normalmente consiste em uma das três opções que podem ser usadas para terapia de reposição hormonal e/ou tratamento de fertilidade.
Sex hormone replacement (testosterone or oestrogen & progesterone).Gonadotropin therapy (medications that replicate the activity of FSH and LH).GnRH pulsatile therapy.Terapia de reposição hormonal
O método e a dose de tratamento variam dependendo do indivíduo que está sendo tratado. O tratamento inicial é normalmente feito com doses mais baixas em pacientes mais jovens, a fim de desenvolver as características sexuais secundárias antes que doses de adultos sejam atingidas.
Para homens com KS/CHH, os tipos de entrega de testosterona incluem manchas diárias, uso diário de gel, cápsulas diárias, injeções subcutâneas ou intramusculares ou implantes de seis meses. Diferentes formulações de testosterona são usadas para garantir que os efeitos anabólicos e androgênicos da testosterona sejam alcançados. Os métodos de entrega de testosterona nasal foram desenvolvidos, mas seu uso no tratamento com KS/CHH não foi formalmente avaliado.
A terapia com gonadotrofina, na forma de injeções de gonadotrofina coriônica humana (HCG), com ou sem o uso de FSH, também podem ser usadas em pacientes do sexo masculino para induzir o desenvolvimento característico sexual secundário, juntamente com a possível indução de fertilidade.
Para as mulheres, a substituição hormonal envolve o uso de estrogênio e progesterona. Em primeiro lugar, o estrogênio é usado na forma de comprimido ou gel para maximizar o desenvolvimento da mama, é utilizado uma combinação de estrogênio e progesterona. Normalmente, a progesterona cíclica é necessária para ajudar a manter o endométrio (revestimento do útero) saudável.
Nos homens, o monitoramento do tratamento normalmente requer a medição da testosterona sérica, inibina B, hematócrito e antígeno específico da próstata (PSA). Se forem utilizadas injeções, são obtidos níveis valiosos para garantir que um nível adequado de testosterona seja alcançado durante todo o ciclo de injeção.
Normalmente, o monitoramento das fêmeas consiste na medição de estrogênio, FSH, LH, inibina B e hormônio anti-Mülleriano (AMH).
A terapia de reposição hormonal padrão normalmente não induzirá a fertilidade em homens ou mulheres, sem crescimento testicular em homens. O tratamento precoce como adolescentes pode ajudar no bem-estar psicológico de pessoas com KS/CHH.
Tratamentos de fertilidade
A terapia com gonadotrofina pode ser usada em pacientes masculinos e femininos, a fim de obter fertilidade para algumas pessoas.
A terapia pulsátil de GnRH também pode ser usada para induzir a fertilidade, especialmente nas mulheres, mas seu uso é limitado a alguns centros de tratamento especializados.
Em homens com KS/CHH, a infertilidade se deve principalmente à falta de produção de espermatozóides dentro dos testículos. A produção de espermatozóides pode ser alcançada através do uso de GnRH administrado por uma bomba de microinfusão ou através do uso de injeções de gonadotrofina (HCG, FSH, HMG). O tempo necessário para alcançar a produção adequada de espermatozóides para a concepção natural variará de pessoa para pessoa. Se os testículos de pré-tratamento forem muito pequenos e houve um histórico de testículos não descidos, pode levar mais tempo para alcançar a produção de espermatozóides. Nesses casos, a tecnologia reprodutiva assistida, como a recuperação de espermatozóides usando a extração de espermatozóides testiculares (Tese) e/ou injeção de esperma intracitoplasmática (ICSI), pode ser necessária.
Nas fêmeas com KS/CHH, a infertilidade se deve principalmente à falta de maturação dos ovos localizados dentro dos ovários. A indução da ovulação pode ser alcançada com terapia pulsátil de GnRH ou alternativamente com injeções de gonadotrofina (HCG, FSH, HMG) fornecidas em intervalos definidos para desencadear a maturação e liberação do ovo para a concepção natural.
Prognóstico
A reversão dos sintomas foi relatada entre 10% a 22% dos casos.
Os casos de reversão foram observados no KS e no ChH normósmico, mas parecem ser menos comuns nos casos de Ks (onde o sentimento de olfato também é afetado). A reversão nem sempre é permanente e as causas genéticas precisas ainda não são totalmente compreendidas.
Epidemiologia
A epidemiologia da síndrome de Kallmann não é bem conhecida. Os estudos individuais incluem um relatório de 1986 que revisou os registros médicos no Exército da Sardenha, que encontrou uma prevalência de 1 em 86.000 homens e um relatório de 2011 da Finlândia, que encontrou uma prevalência de 1: 30.000 para homens e 1: 125.000 para mulheres.
A síndrome de Kallmann ocorre cerca de 4 vezes mais frequentemente em homens do que mulheres, mas é apenas 2,5 vezes mais comum entre os homens em casos familiares.
História

A síndrome de Kallmann foi descrita pela primeira vez pelo nome em um artigo publicado em 1944 por Franz Josef Kallmann, um geneticista alemão-americano. A ligação entre a anosmia e o hipogonadismo já havia sido observada pelo médico espanhol Aureliano Maestre de San Juan em 1856. Na década de 1950, de Morsier e Gauthier relataram a ausência parcial ou completa da lâmpada olfativa no cérebro de homens com hipogonadismo.
Terminologia
A terminologia usada ao descrever casos de HH variam e pode incluir: [citação necessária]
GnRH deficiencycongenital hypogonadotropic hypogonadism (CHH)idiopathic/isolated hypogonadotropic hypogonadism (IHH)normosmic hypogonadotropic hypogonadism (nHH)hypothalamic hypogonadismolfacto-genital syndromePesquisar
Kisspeptina é uma proteína que regula a liberação de GnRH do hipotálamo, que por sua vez regula a liberação de LH e, em menor grau, FSH da glândula pituitária anterior. Kisspeptina e seu receptor associado Kiss1r são conhecidos por estarem envolvidos na regulamentação da puberdade. Estudos demonstraram que há potencial para que a Kisspeptina seja usada no diagnóstico e tratamento de certos casos de síndrome de Kallmann e CHH.